



Group leader: Ferdinando Di Cunto

The development of the central nervous system requires careful processes of cell division and differentiation. An alteration of these events can severely reduce the number of neurons and impair brain function, causing severe symptoms such as intellectual disability, movement disorders and epilepsy.
Our group studies how the proliferation, vitality and differentiation of neurons can be altered by different genetic mutations, resulting in syndromes known as microcephaly. Furthermore, we seek to understand how the molecular mechanisms of microcephaly can provide useful therapeutic targets to fight brain tumors, in particular those affecting the pediatric age, such as medulloblastoma.
The human brain is composed of approximately 90 billion neurons, which are generated during embryonic life starting from many different types of neural stem cells, whose proliferation is extremely well organized in space and time. Indeed, stem cells' proliferation is very intense during early brain development, but chases almost completely in post-natal life. If too few neurons are produced or too many neurons die during development, the brain volume can be very compromised, a condition commonly known as microcephaly.
Although a significantly reduced brain volume can be compatible with normal brain function and intelligence, microcephaly is frequently associated with strongly invalidating symptoms, such as intellectual disability, epilepsy and cerebral palsy. Microcephaly can be the result of rare genetic disorders, mostly characterized by autosomal recessive inheritance. Even more frequently, it is produced by environmental factors, such as hypoxia, drugs and alcohol exposure or infectious agents, such as Rubella, Toxoplasmosis, Cytomegalovirus or Zyka virus. Research conducted in the last decade has shown that all these conditions may affect common molecular pathways, regulating genome stability, cell proliferation, cell survival and determination of cell identity.
The main focus of our group is to understand how these common mechanisms are related and to develop new strategies that may prevent neuron loss during development, thereby attenuating the consequences of microcephaly-causing insults. We are also studying how abnormal development may lead to abnormal neural circuits, not only in genetic microcephaly, but also in Down syndrome.
Implementation of models for the study of neurodegenerative diseases and neurodevelopment
Despite the constant increase of the frequency of neurological disorders of genetic origin, not many solutions for a reliable preclinical diagnosis and for development of effective treatments have been identified. One of the most limiting factors for these activities is the scarceness of suitable experimental models, capable of connecting clinical and biological studies. This problem has become particularly evident after the strong advance of the possibility to genetically study patients, offered by modern sequencing technologies. With the aim of increasing the integration of NICO with clinical research centers, we aim at implementing in our institute high-throughput models for the functional study of mutations: in vitro, through immortalized cell lines; in vivo, using the genetically treatable model Caenorhabditiselegans.
Specifically, our research has the aim of clarifying:
-
How mutations in Citron kinase lead to microcephaly.
Citron kinase (CITK) is a protein that regulates the very last step of the cell division cycle, commonly known as cytokinesis, responsible for splitting a dividing cell into the two daughters. We have first discovered that the complete loss of this protein leads to a severe form of microcephaly in experimental models. Recently, genome sequencing of patients affected by severe microcephaly has identified mutations in the Citron kinase gene, demonstrating its role in pathogenesis of human microcephaly. We know that the neural progenitors of individual carrying CITK mutations fail to divide and undergo programmed cell death, leading to strong reduction of the neuron number. We are working to clarify how cytokinesis failure may lead to cell death, and whether other mechanisms may be responsible for inducing apoptosis or reducing neuron numbers. Our last discovery on this topic is that CITK may functionally cooperate with ASPM, the gene most frequently mutated in genetic microcephaly. Together, the two proteins may control the orientation of mitotic spindle and the frequency of symmetric and asymmetric cell division, which is known to control neuron numbers independently from cell death.
-
What are the molecular consequences of CITK loss.
CITK was originally identified as a protein important for remodeling the actin cytoskeleton. We have recently found that it may be even more important to regulate the stability of microtubules, and that this function is crucial for completing cytokinesis and for spindle orientation.
-
Neuronal alterations in Down syndrome.
Down syndrome (DS) is a multi-genic disorder produced by trisomy of Chromosome (Chr.) 21 and principally characterized by intellectual disability (ID), which also represents the most invalidating manifestation of the disease. However, the causative events that alter neuronal circuitry within the cortex remain unknown. We are addressing the possibility that at the basis of the modified connections might be cortical migration and lamination deficits. In particular, we are focus our attention on the role TTC3, a gene located in the region of Chr. 21 believed to play the strongest role in determining intellectual disability.
-
CITK as a possible target for cancer therapy.
We are addressing the possibility that the function of CITK may be essential for proliferation in medulloblastomas, devastating brain tumors of the infancy that urgently require the development of new therapies.
News
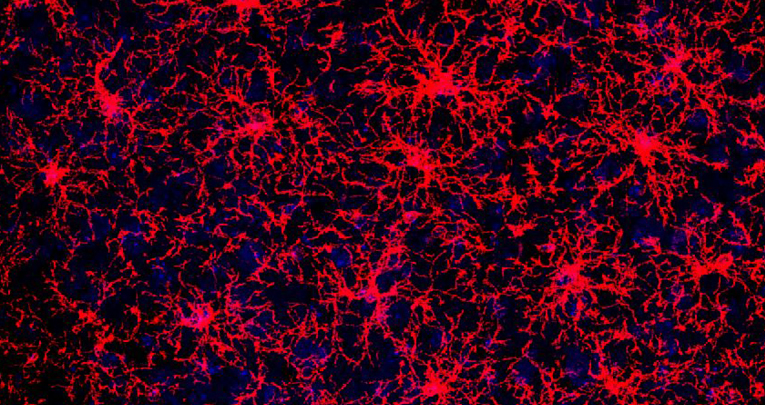
Not all oligodendrocyte precursors are created equal
Nature Communication , 28 April 2022
Molecular and functional heterogeneity in dorsal and ventral oligodendrocyte progenitor cells of the mouse forebrain in response to DNA damage.
Enrica Boda, Martina Lorenzati, Roberta Parolisi, Brian Harding, Gianmarco Pallavicini, Luca Bonfanti, Amanda Moccia, Stephanie Bielas, Ferdinando Di Cunto, Annalisa Buffo
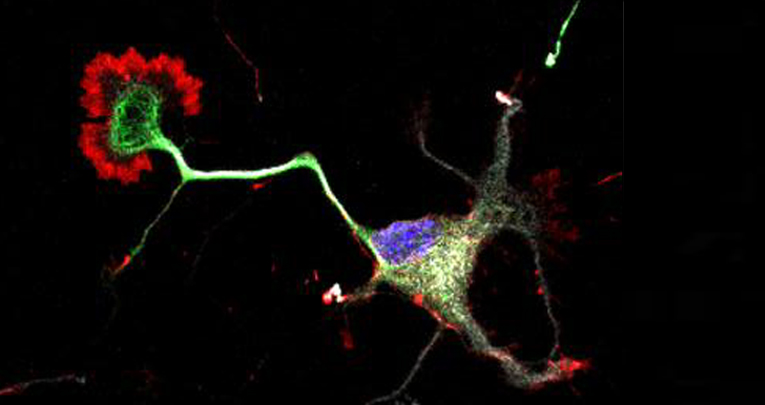
miR-7b-3p Exerts a Dual Role After Spinal Cord Injury, by Supporting Plasticity and Neuroprotection at Cortical Level
Frontiers in Molecular Biosciences , 31 March 2021
Ghibaudi M, Boido M, Green D, Signorino E, Berto GE, Pourshayesteh S, Singh A, Di Cunto F, Dalmay T, Vercelli A
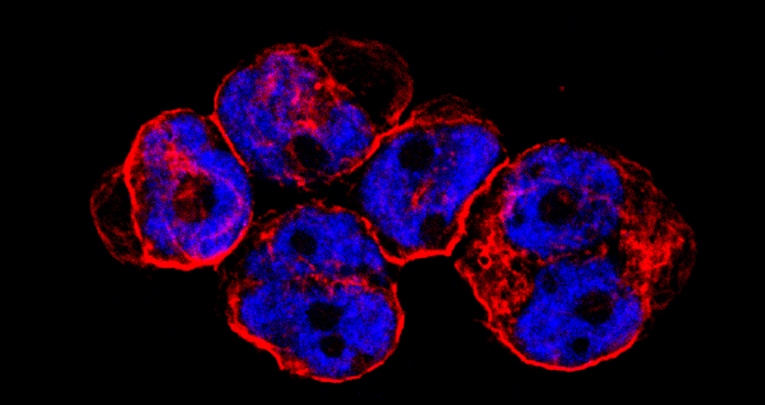
CITK loss inhibits growth of Group 3 and 4 medulloblastoma cells and sensitizes them to DNA-damaging agents.
Cancers , 26 February 2020
G Pallavicini, G Iegiani, G Berto, E Calamia, E Trevisiol, A Veltri, S Allis and F Di Cunto
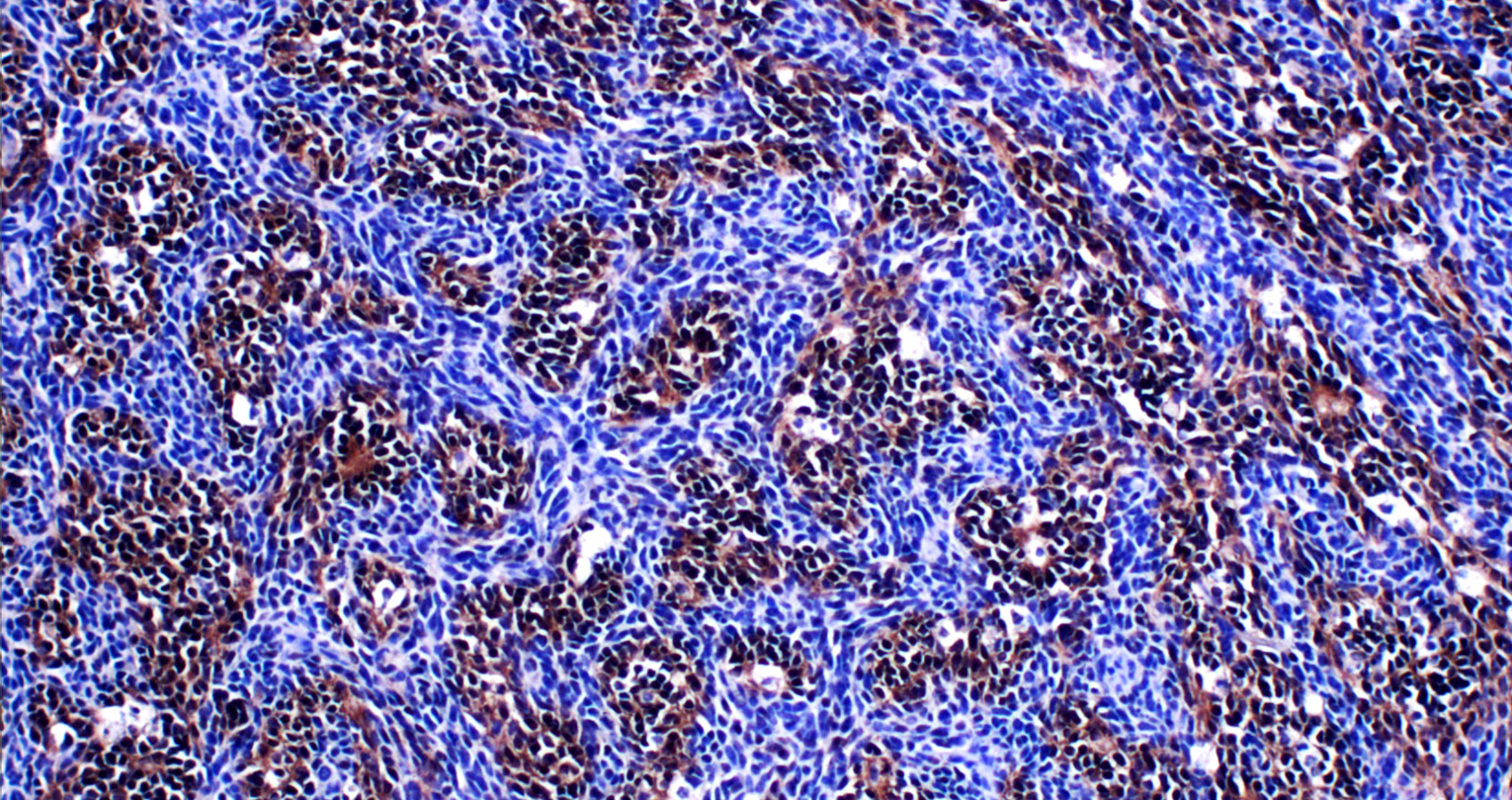
Inactivation of citron kinase inhibits medulloblastoma progression by inducing apoptosis and cell senescence.
Cancer Research
, June 2018
Pallavicini G, Sgro F, Garello F, Falcone M, Bitonto V, Berto GE, Bianchi FT, Gai M, Chiotto AM, Filippi M, Cutrin JC, Ala U, Terreno E, Turco E, Di Cunto F