



Gruppo guidato da Annalisa Buffo


Annalisa Buffo
Contacts:
Tel +39 011 670 6614
Fax +39 011 670 6621
e-mail: annalisa.buffo@unito.it
Born in Turin on 25.12.1967 - Italian.
Education
1991 MBio summa cum laude, University of Turin
1992-1993 Post-lauream apprenticeship at the II Neurological Clinic, Faculty of Medicine, University of Turin, Italy
1994 Professional certificate for Biologists, University of Turin, Italy
1998 Research Doctorate in Neurological Sciences, University of Turin, Italy
1994-1999 Research stays at the Rudolf Magnus Institute, University of Utrecht, Netherlands Institute for Brain Research, Amsterdam, and University of Tuebingen, Germany
Former and current positions
1992-1998 Research fellow, University of Turin
1998-2000 Postdoctoral fellow, Department of Physiology, University of Turin
2001-2016 Tenured Assistant Professor, University of Turin
2004, 2005 Visiting Researcher at the Helmholtz Zentrum (GSF, now Deutsches Forschungszentrum für Gesundheit und Umwelt, GmbH) and Ludwig Maximilans University, Munich, Germany
2017-present Associate Professor in Physiology, University of Turin
2016-present Co-founder of the Start-up S&P Brain (www.spbrain.com)
2019-present Deputy Director of the Neuroscience Institute Cavalieri Ottolenghi
2018 Editorial Board member of Scientific Reports
Main research interests
During the last years my research has focussed on the role of glia and progenitor cells in brain plasticity and repair, and on the implementation of cell replacement approaches in conjunction with training protocols to promote functional recovery in CNS diseases.
We believe that specific issues regarding glia and neural progenitors are particularly promising to both unveil new keys to the understanding of physiology, and accomplish therapeutic actions. We have revealed that a fundamental mechanisms oligodendrocyte progenitor self-maintenance is asymmetric cell division (Boda et al., Glia 2015), and provided among the first demonstrations of oligodendrocyte alterations and myelin abnormalities in Alzheimer disease (Behrendt et al., Glia 2013) and Autism models (Bertero et al., Brain 2018). We also have contributed to set up first in vitro rodent and human neural models of leukodystrophy (ADLD) to validate an allele-specific silencing therapeutic strategy (Giorgio et al., Brain, 2019).
As for astroglia, we have made important contributions on the development of astrocyte heterogeneity, and on how it differentially impacts on CNS functions is unknown. We addressed this topic by studying cerebellar morphogenesis, which lead to the first demonstration that an ontogenetic program, tightly regulated in space and time, determines astrocyte heterogeneity (Cerrato et al., PLoS Biol 2018). In parallel studies, we identified a functional requirement for SOX2 in postnatal Bergmann astrocyte differentiation and functions, of relevance for ataxia in mouse mutants, and in human patients (Cerrato et al., Glia 2018).
Further, we disclosed the first lineage contiguity between astrocytes and interneurons, demonstrating that bipotent astroglial-like progenitors give rise to both interneurons and white matter astrocytes (Parmigiani et al., J Neurosci 2015). One further key aspect of astrocyte heterogeneity is the link between adult parenchymal astrocytes and neural stem cells. We contributed to show that after excitotoxic lesion subsets of striatal astrocytes undergo a spontaneous neurogenic activation leading to the local generation of a large amount of neuroblasts (Nato et al., Development 2015). We are now exploring the underlying molecular mechanisms of both diversification of astrocyte types, and activation of neurogenic properties.
In respect to strategies to promote functional recovery in neurodegenerative models, we focused on a model for progressive ataxia caused by overactivation of autophagy leading to selective loss of Purkinje neurons. We showed that preventive transplantation does not alleviate the ataxic symptoms whereas preventive motor training increases Purkinje neuron survival with positive consequences on both cerebellar circuit alterations and the motor phenotype suggesting the use of preventive motor exercise in inherited ataxia to promote a delay in the progression of the disease (Fucà et al., Neurobiol Dis 2018). We translate this knowledge in parallel studies addressing the efficacy of hPSC-based cell replacement approaches in rodent models of Huntington’s disease.
Role in research
Principal investigator
Il Neurone Immortale?
scopri il progetto di ricerca

Publications:
> Publicationslist.org
> PubMed.gov
> ORCID
S&P BRAIN
Services and Products for preclinical proof of concepts
Our spinoff provides scientific expertise, animal models, equipment and facilities to pharmaceutical, biotechnology, and medical device Companies and to Research Centers for proof–of-concept or pilot in vivo studies.
News
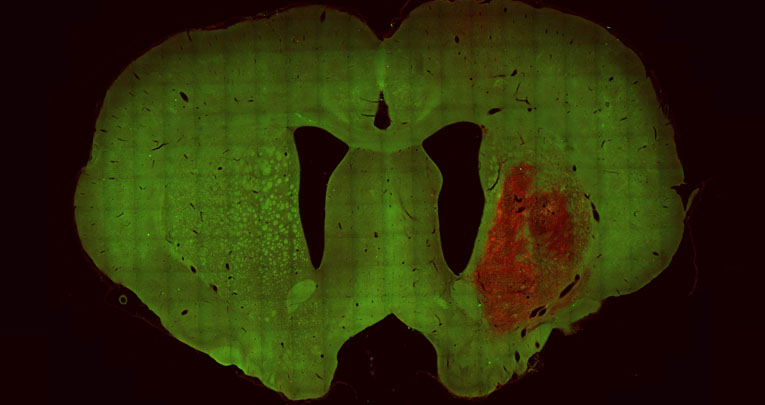
Malattia di Huntington: il valore terapeutico del trapianto di neuroni derivati da cellule staminali umane
A oggi non esistono trattamenti efficaci in grado di arrestare la neurodegenerazione e quindi la progressione della malattia di Huntington (HD). Nello studio pubblicato su Stem Cell Research & Therapy le ricercatrici del NICO - guidate dalla prof.ssa Annalisa Buffo - dimostrano che il trapianto di neuroni striatali derivati da cellule staminali embrionali umane potrebbe avere un valore terapeutico per l’HD.
Lo studio - realizzato in collaborazione con il laboratorio della prof.ssa Elena Cattaneo dell’Università Statale di Milano - è finanziato dai consorzi europei Neurostemcell Repair e Neurostemcell Reconstruct.
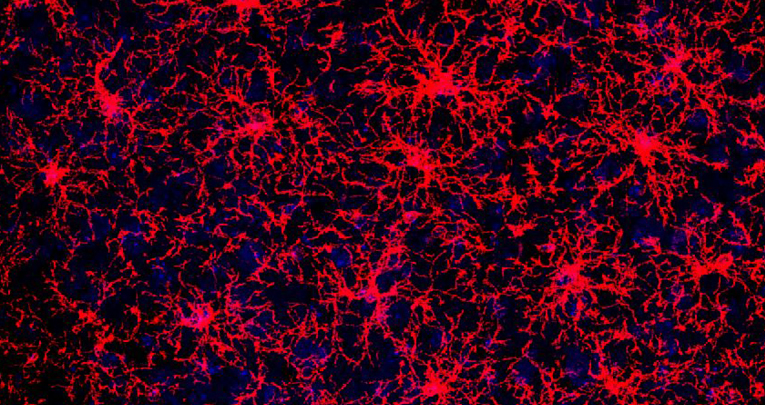
L'eredità nascosta delle cellule: nuova luce sui progenitori degli oligodendrociti
Chiarita la relazione fra l’eterogeneità di queste cellule del Sistema Nervoso Centrale e la loro risposta al danno al DNA, responsabile dell’invecchiamento delle cellule e coinvolto in molte patologie che colpiscono il cervello. Lo studio - guidato dalla prof.ssa Enrica Boda del NICO Università di Torino - pubblicato sulla prestigiosa rivista Nature Communication.